18 May 2021: Original Paper
Diagnostic Dilemma and Treatment Outcome in Acute Liver Failure Due to Wilson’s Disease
Rafał Stankiewicz1ABCDEF*, Waldemar Patkowski1DG, Krzysztof Zieniewicz1ADGDOI: 10.12659/AOT.930146
Ann Transplant 2021; 26:e930146






Abstract
BACKGROUND: Wilson’s disease (WD) manifesting as acute liver failure (ALF) is a life-threatening condition, and spontaneous recovery is rare. Diagnostic scores like the alkaline phosphatase elevation/total bilirubin elevation ratio and aspartate aminotransferase/alanine aminotransferase ratio can distinguish WD from other ALF etiologies. Liver transplantation plays a major role in treating these patients, and the revised Wilson Index is useful in patient selection for this procedure. The aim of this study was to evaluate diagnostic scores, treatments, and outcomes of a large cohort of patients with WD-ALF.
MATERIAL AND METHODS: Twenty adult patients of a historical cohort admitted from January 2001 to December 2017 were prospectively observed. Demographic, clinical, laboratory, and radiology data, and treatment, time on the waiting list for liver transplantation, and outcomes were recorded.
RESULTS: No diagnostic laboratory scores were 100% positive in patients with WD-ALF. Cut-off values for the alkaline phosphatase/total bilirubin ratio and aspartate aminotransferase/alanine aminotransferase ratio were met by 65.0% and 80.0% of patients, respectively. All patients met at least 1 criterion for high risk of death (Nazer or revised Wilson Index) and qualified for liver transplantation. In 9 patients, albumin dialysis was used before surgery. Survival after liver transplantation was 85.0% and 74.4% after 1 month and 1 year, respectively.
CONCLUSIONS: Further research on a novel diagnostic score in WD-ALF is warranted. Adult patients suspected to have WD as the cause of ALF should be treated in the referral liver transplantation unit. Liver transplantation makes long-term survival possible for patients with this critical illness.
Keywords: Hepatolenticular Degeneration, Liver Failure, Acute, Liver Transplantation, Liver, Artificial, Survival, Adolescent, End stage liver disease, Severity of Illness Index, young adult
Background
Wilson’s disease (WD) is an autosomal recessive disorder which leads to the accumulation of copper, particularly in the liver and brain, due to the mutation of the
Previous studies on patients with WD-ALF comprise small cohorts of pediatric and adult patients with decompensated chronic liver failure and ALF. The objective of this study was to report our experience in the diagnosis and treatment of ALF due to WD in adults.
Material and Methods
Twenty adult patients of a historical cohort with WD-ALF who were admitted from January 2001 to December 2017 were prospectively observed. All patients met criteria of ALF diagnosis: international normalized ratio (INR) ≥1.5 and encephalopathy. No history of liver disease was present up to 26 weeks prior to first admission [9]. WD as a cause of ALF was diagnosed after the admittance to our center in most of the patients. The diagnosis of WD was based on the following criteria: low serum ceruloplasmin level, elevated 24-h urinary copper excretion, increased serum “free” copper, presence of K-F rings by slit lamp examination, and, from 2008 onward, an ALP/TB ratio <4 and AST/ALT ratio >2.2 [5]. For study purposes, patients treated before 2008 had an ALP/TB ratio and AST/ALT ratio calculated retrospectively. Genetic evaluation was not routinely available during the whole study period. Other causes of ALF such as toxins, drugs or mushroom poisoning, viral infection, and autoimmune hepatitis were excluded.
The patients’ medical records were analyzed for demographic data (including age and sex), clinical data (grade of encephalopathy, time between the start of jaundice and the development of encephalopathy to classify it as hyperacute, acute, or subacute [10]), laboratory data, radiology data (abdominal Doppler ultrasound and/or computed tomography), treatment, time on the waiting list for liver transplantation, and outcomes.
Selection for liver transplantation was based on the King’s College criteria for non-paracetamol causes. The Nazer index and the revised Wilson Index, from 2005 onward, were supportive in the decision-making process. The King’s College criteria are defined as an INR over 6.5 or at least 3 of the following criteria: drug toxicity; age below 10 years old or over 40 years old; an interval between jaundice and encephalopathy over 7 days; and INR level over 3.5 and serum bilirubin over 17.5 mg/dL [11]. The Nazer index (0–4 points) grades prolongation in prothrombin time, AST activity, and serum bilirubin. A score of 7 or higher predicts a fatal outcome [7]. The revised Wilson Index (0–4 points) grades serum bilirubin, AST, INR, white blood cell count, and serum albumin [8]. A score ≥11 is associated with a high probability of death without liver transplantation. For study purposes, patients treated before 2005 had a revised Wilson Index calculated retrospectively.
At the same time as they were listed for liver transplantation, patients were qualified for albumin dialysis: fractionated plasma separation and absorption with high-flux dialysis (FPSA, Prometheus®). Selection for additional treatment required at least 3 of the following: encephalopathy grade 3/4; serum bilirubin >15 mg/dL; increase of transaminases over 50% in consecutive tests; serum ammonia level over standard range; and serum creatinine or urea levels over standard range. FPSA was performed once daily for a maximum of 3 procedures, or fewer if a liver donor was available. In patients with no availability of liver transplantation, the FPSA was not continued after 3 procedures, according to treatment protocol [12].
Explants of native livers after liver transplantation were routinely examined with hematoxylin and eosin staining. In addition, rhodamine and orcein stains were performed to identify copper granules and copper-binding protein, respectively.
Institutional review board permission was obtained for reviewing the patient clinical charts for research purposes.
Laboratory data are presented as the mean and standard deviation (SD). The survival time after liver transplantation was estimated with the Kaplan-Meier method, and the log-rank test was used to compare survival curves.
Results
CLINICAL FEATURES:
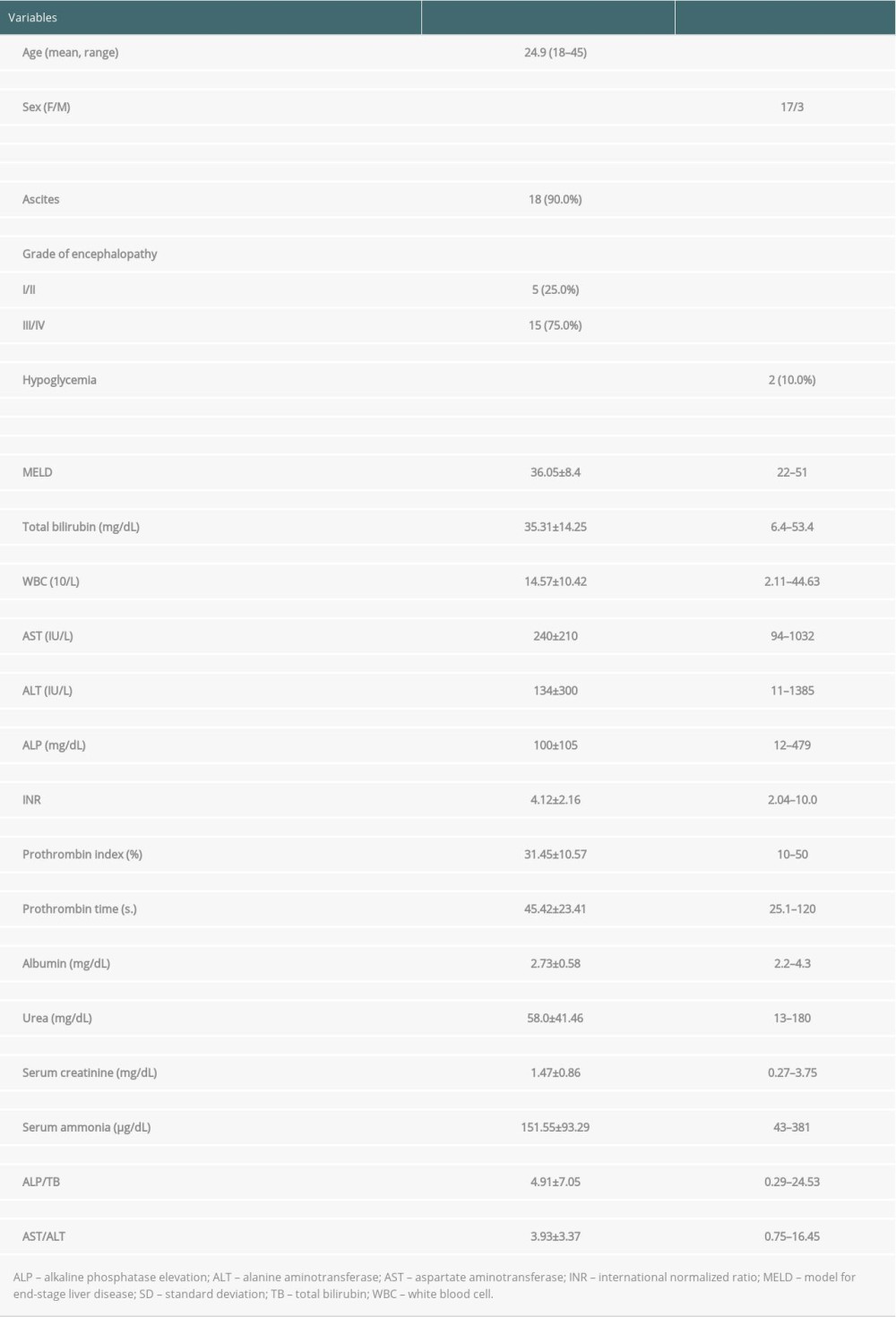
The median age of the group was 21 (range, 18–45) years, and only 3 patients were men. The mean time interval between the first manifestation and admission was 30 (range, 1–84) days. On admission, jaundice and encephalopathy were present in all patients, and ascites was present in 18 (90.0%) patients. Renal insufficiency, defined as serum creatinine level over baseline (>1.2 mg/dL) with decrease in urine output <50 mL/h, was present in 11 (55.0%) patients. One patient had K-F rings, 9 (45.0%) had decreased ceruloplasmin concentration, 5 (25.0%) had abnormal values of serum copper, and 9 (45.0%) had elevated urinary copper excretion. Thirteen (65.0%) patients met the cut-off for the ALP/TB ratio and 16 (80.0%) met the cut-off for the AST/ALT ratio. Two patients did not fulfill any of the laboratory criteria for WD-ALF, but 1 had K-F rings, and in both cases the diagnosis was confirmed in histopathology of the explanted livers. The clinical and laboratory data are shown in Table 1. Ten patients (50.0%) had radiological (ultrasound or computer tomography) signs of cirrhosis or portal hypertension.
TREATMENT:
All patients proceeded to liver transplantation after a median of 6 (range 1–28) days from admission. Thirteen (65.0%) patients met the King’s College criteria for non-paracetamol causes. The mean model for end-stage liver disease was 36.05 (range 22–51). Eighteen (90.0%) patients reached or exceeded the cut-off value of the Nazer index for a high risk of death (2 patients had 6 points on the Nazer index). The cut-off value of the revised Wilson Index was met by 18 (90.0%) patients (1 patient had 10 points and 1 patient had 9 points). All patients met at least 1 criterion for a high risk of death without liver transplantation.
Nine (47.4%) patients were treated with FPSA (1 patient was treated before Prometheus® was available). Five patients had 3 procedures, 1 patient had 2 procedures, and 3 patients had 1 procedure of the albumin dialysis.
OUTCOMES:
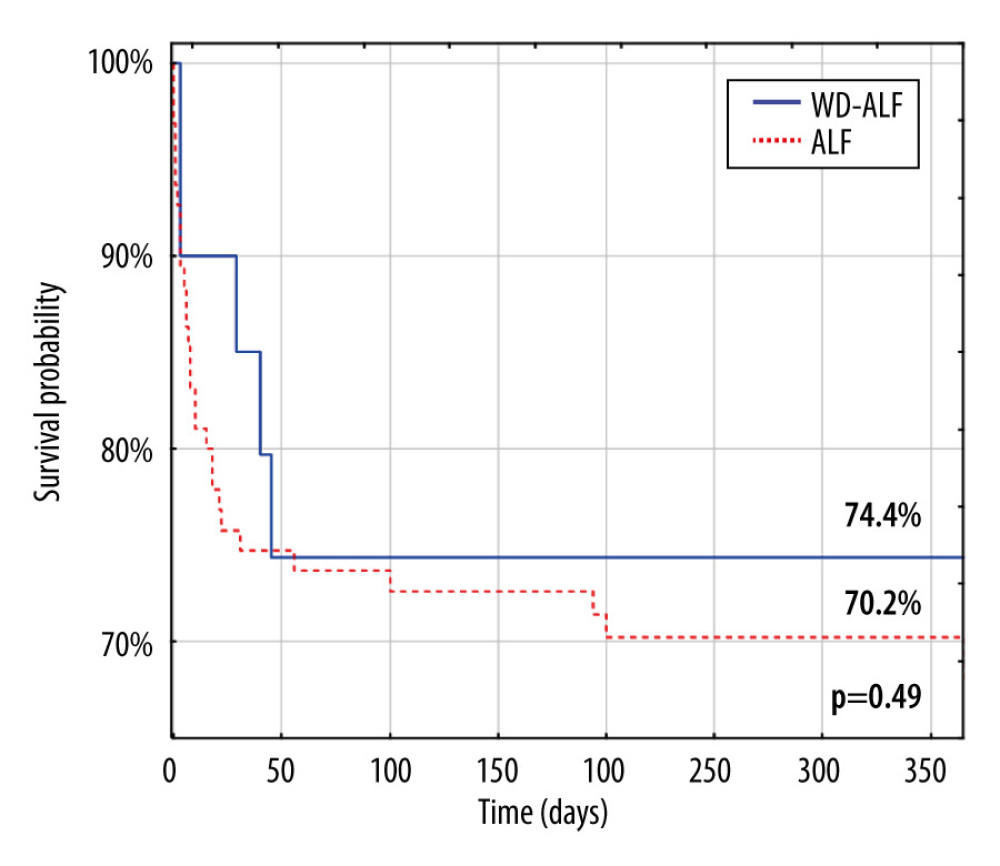
Survival after liver transplantation was 85.0% and 74.4% after 1 month and 1 year, respectively. Three patients died in the early posttransplant period (median, 3 [range 3–29] days); 2 cases were due to multiorgan failure and 1 case was due to primary nonfunction. There was 1 liver retransplantation 12 days after the primary operation, owing to hepatic artery thrombosis.
Copper deposits were identified in 9 (45.0%) explanted livers, confirming the diagnosis of WD-ALF.
Additionally, survival of the WD-ALF cohort was compared with non-WD-ALF patients (n=95) treated with liver transplantation during the same period (Figure 1).
Discussion
The diagnosis of WD-ALF is vitally important because of the high mortality without liver transplantation and implications for family members. It has been established that the diagnosis of acute presentation is more challenging than that in the chronic setting. For example, K-F rings are detectable in only up to 50% of patients with WD, and obtaining a slit lamp examination in patients in critical condition can be difficult. Serum copper is neither specific nor sensitive for the diagnosis of WD-ALF. Reduction of urine output due to impaired renal function in up to 40% of patients with ALF and delay in obtaining results from reference laboratories limits the potential utility of urinary copper excretion [6,13,14]. Our findings support these observations. Only 1 patient in our study had K-F rings, serum copper was elevated in 25.0% of patients, and urinary copper was elevated in 45.0% of patients.
Late-onset presentation of WD as a cause of ALF is rare. There are few cases of first presentation of WD after the age of 40 years [15]. In the presented cohort, 1 woman was diagnosed at the age of 45 with K-F rings and was treated with a successful liver transplantation.
Liver biopsy to determine hepatic copper levels is not practical owing to the high risk of bleeding in patients with ALF. Furthermore, in the study by Korman et al, not every case of histochemical staining confirmed copper deposits in the histologic evaluation of 7 available liver samples [6]. This is in accordance with our findings, where copper deposits were found in the explanted livers of only 45.0% of patients.
For the rapid diagnosis of WD-ALF, 2 index scores are in use. The ALP/TB ratio is based on the observation that in the course of acute presentation of WD, the ALP activity decreases, and bilirubin concentration increases [14]. The AST/ALT ratio reflects the concept of mitochondrial injury in fulminant WD [16]. The cut-off values for both indexes were reevaluated by Korman et al in 2008. They found a sensitivity of 94% and 94% and specificity of 96% and 86% for the ALP/TB ratio <4 and AST/ALT ratio >2.2, respectively, in the diagnosis of WD-ALF. The same authors showed that by first screening patients with the ALP/TB ratio and then sequentially screening with the AST/ALT ratio, the sensitivity and specificity increase to 100% [6]. In the present study, 65% of patients had a positive ALP/TB ratio and 80% of patients had a positive AST/ALT ratio. By using the sequential method, the positive diagnostic value was 90%.
Difficulties in creating a diagnostic algorithm for WD-ALF are reflected in recently published guidelines. The American Association for the Study of Liver Disease (AASLD), European Association for the Study of the Liver (EASL), and European Society for Pediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN) propose various diagnostic tests and cut-off values. For example, in the AASLD guidelines, the cut-off for the ALP/TB ratio for WD-ALF diagnosis is less than 2, while the EASL and ESPGHAN specify a ratio of ALP to TB of less than 4 and 1, respectively [17].
In 2020, Gungor et al constructed a scoring system to distinguish between WD-ALF and non-WD-ALF in a pediatric population [18]. Based on AST, ALT, ALP, AST/ALT ratio, uric acid, and hemoglobin concentrations, their scoring system had a sensitivity of 87.5% and specificity of 86.7%. Its application should be further assessed, and its utility in adults needs to be studied. Unfortunately, this could not be done in the present adult population because of the lack of availability of uric acid in the historic cohort. Also, new cut-off points must be established for adults, owing to age-related changes in enzyme activity.
In 1991, Berman et al reported uniformly fatal outcomes in patients with WD-ALF, and not until 2007 were the first cases of survival without liver transplantation published [19,20]. Patients surviving ALF due to WD are mostly from the pediatric population [18,20–23]. Scarce data exist of adults with WD-ALF who survived without liver transplantation; 1 case was published by Damsgaard et al in 2019. The 27-year-old male patient with a high revised Wilson Index (16 points) was treated successfully with high-volume plasma exchange and penicillamine [24].
Czuprynska et al reported a cohort of 14 adult patients in a Polish population with WD-ALF. No recovery without liver transplantation was observed, and the 1-year survival rate was 85.7%. No albumin dialysis was used in the treatment [25]. A study from Italy reported an 88% 1-year survival rate of adult patients after liver transplantation for WD, but the main indication was chronic liver failure (n=25), and only 1 patient had ALF [26].
Our study results were in accordance with this previous data. No patient with WD-ALF survived without liver transplantation. Survival after liver transplantation in the presented group of patients was significantly better than that in the general population of patients with ALF: 74.4% vs 70.2% after 1 month (
Albumin dialysis (FPSA) proved to be an effective bridging treatment in this study, prolonging waiting time to liver transplantation.
Conclusions
Because of the difficulty of diagnosis and the high risk of fatal outcome, adult patients suspected of having WD as the cause of ALF should be treated in a referral liver transplantation unit, with access to albumin dialysis if possible. The excellent outcomes after liver transplantation make long-term survival possible for patients with this critical illness.
References
1. Frydman M, Genetic aspects of Wilson’s disease: J Gastroenterol Hepatol, 1990; 5; 483-90
2. Scheinberg IH, Sternlieb I, Wilson’s disease: Major Problems in Internal Medicine, 1984; 23; 25-35, Philadelphia, PA, WB Saunders
3. Członkowska A, Litwin T, Dusek P, Wilson disease: Nat Rev Dis Primers, 2018; 4(1); 21
4. Ostapowicz G, Fontana RJ, Schiødt FV, Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States: Ann Intern Med, 2002; 137(12); 947-54
5. European Association for Study of Liver, EASL Clinical Practice Guidelines: Wilson’s disease: J Hepatol, 2012; 56(3); 671-85
6. Korman JD, Volenberg I, Balko J, Screening for Wilson disease in acute liver failure: A comparison of currently available diagnostic tests: Hepatology, 2008; 48(4); 1167-74
7. Nazer H, Ede RJ, Mowat AP, Williams R, Wilson’s disease: Clinical presentation and use of prognostic index: Gut, 1986; 27(11); 1377-81
8. Dhawan A, Taylor RM, Cheeseman P, Wilson’s disease in children: 37-year experience and revised King’s score for liver transplantation: Liver Transpl, 2005; 11(4); 441-48
9. European Association for the Study of the Liver, Clinical practice guidelines panel, Wendon, J; Panel members, Cordoba J, Dhawan A, Larsen FS, et al. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure: J Hepatol, 2017; 66(5); 1047-81
10. O’Grady JG, Schalm SW, Williams R, Acute liver failure: redefining the syndromes [published erratum appears in Lancet 1993;342:1000]: Lancet, 1993; 342; 273-75
11. O’Grady JG, Alexander GJ, Hayllar KM, Williams R, Early indicators of prognosis in fulminant hepatic failure: Gastroenterology, 1989; 97; 439-45
12. Stankiewicz R, Lewandowski Z, Kotulski M, Effectiveness of fractionated plasma separation and absorption as a treatment for amanita phalloides poisoning: Ann Transplant, 2016; 21; 428-32
13. Steindl P, Ferenci P, Dienes HP, Wilson’s disease in patients presenting with liver disease: A diagnostic challenge: Gastroenterology, 1997; 113; 212-18
14. Shaver WA, Bhatt H, Combes B, Low serum alkaline phosphatase activity in Wilson’s disease: Hepatology, 1986; 6; 859-63
15. Shribman S, Webb G, Taylor R, Liver transplantation for late-onset presentations of acute liver failure in Wilson’s disease: The UK experience over 2 decades: JHEP Rep, 2020; 2(3); 100096
16. Schilsky ML, Diagnosis and treatment of Wilson’s disease: Pediatr Transplant, 2002; 6; 15-19
17. Saroli Palumbo C, Schilsky ML, Clinical practice guidelines in Wilson disease: Ann Transl Med, 2019; 7(Suppl 2); S65
18. Gungor S, Semimoglu MA, Bag HG, Varol FI, Is it possible to diagnose fulminant Wilson’s disease with simple laboratory tests?: Liver Int, 2020; 40(1); 155-62
19. Berman DH, Leventhal RI, Gavaler JS, Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure: Gastroenterology, 1991; 100(4); 1129-34
20. Eisenbach C, Sieg O, Stremmel W, Diagnostic criteria for acute liver failure due to Wilson disease: World J Gastroenterol, 2007; 13(11); 1711-14
21. Markiewicz-Kijewska M, Szymczak M, Ismail H, Liver transplantation for fulminant Wilson’s disease in children: Ann Transplant, 2008; 13(2); 28-31
22. Kido J, Matsumoto S, Sakamoto R, Recovery of severe acute liver failure without transplantation in patients with Wilson disease: Pediatr Transplant, 2018; 22(8); e13292
23. Mainardi V, Rando K, Valverde M, Acute liver failure due to wilson disease: Eight years of the National Liver Transplant Program in Uruguay: Ann Hepatol, 2019; 18(1); 187-92
24. Damsgaard J, Larsen FS, Ytting H, Reversal of acute liver failure due to wilson disease by a regimen of high-volume plasma exchange and penicillamine: Hepatology, 2019; 69(4); 1835-37
25. Czuprynska MM, Wawrzynowicz-Syczewska M, Jureczko L, Preemptive administration of recombinant factor VII (rVIIa) in patients transplanted due to fulminant Wilson’s disease: Ann Transplant, 2010; 15(3); 7-10
26. Ferrarese A, Morelli MC, Carrai P, Outcomes of liver transplant for adults with Wilson’s Disease: Liver Transpl, 2020; 26(4); 507-16
In Press
18 Mar 2024 : Original article
Does Antibiotic Use Increase the Risk of Post-Transplantation Diabetes Mellitus? A Retrospective Study of R...Ann Transplant In Press; DOI: 10.12659/AOT.943282
20 Mar 2024 : Original article
Transplant Nephrectomy: A Comparative Study of Timing and Techniques in a Single InstitutionAnn Transplant In Press; DOI: 10.12659/AOT.942252
28 Mar 2024 : Original article
Association Between FEV₁ Decline Rate and Mortality in Long-Term Follow-Up of a 21-Patient Pilot Clinical T...Ann Transplant In Press; DOI: 10.12659/AOT.942823
02 Apr 2024 : Original article
Liver Transplantation from Brain-Dead Donors with Hepatitis B or C in South Korea: A 2014-2020 Korean Organ...Ann Transplant In Press; DOI: 10.12659/AOT.943588
Most Viewed Current Articles
05 Apr 2022 : Original article
Impact of Statins on Hepatocellular Carcinoma Recurrence After Living-Donor Liver TransplantationDOI :10.12659/AOT.935604
Ann Transplant 2022; 27:e935604
12 Jan 2022 : Original article
Risk Factors for Developing BK Virus-Associated Nephropathy: A Single-Center Retrospective Cohort Study of ...DOI :10.12659/AOT.934738
Ann Transplant 2022; 27:e934738
22 Nov 2022 : Original article
Long-Term Effects of Everolimus-Facilitated Tacrolimus Reduction in Living-Donor Liver Transplant Recipient...DOI :10.12659/AOT.937988
Ann Transplant 2022; 27:e937988
15 Mar 2022 : Case report
Combined Liver, Pancreas-Duodenum, and Kidney Transplantation for Patients with Hepatitis B Cirrhosis, Urem...DOI :10.12659/AOT.935860
Ann Transplant 2022; 27:e935860